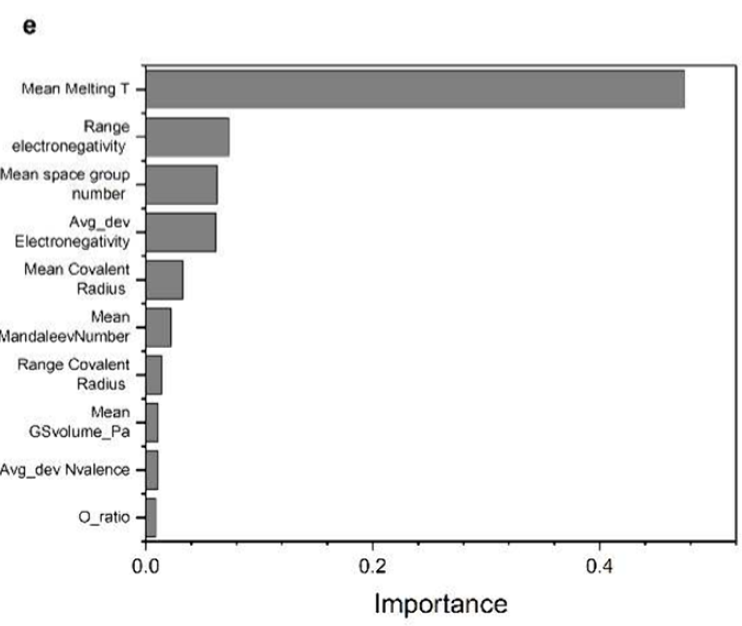
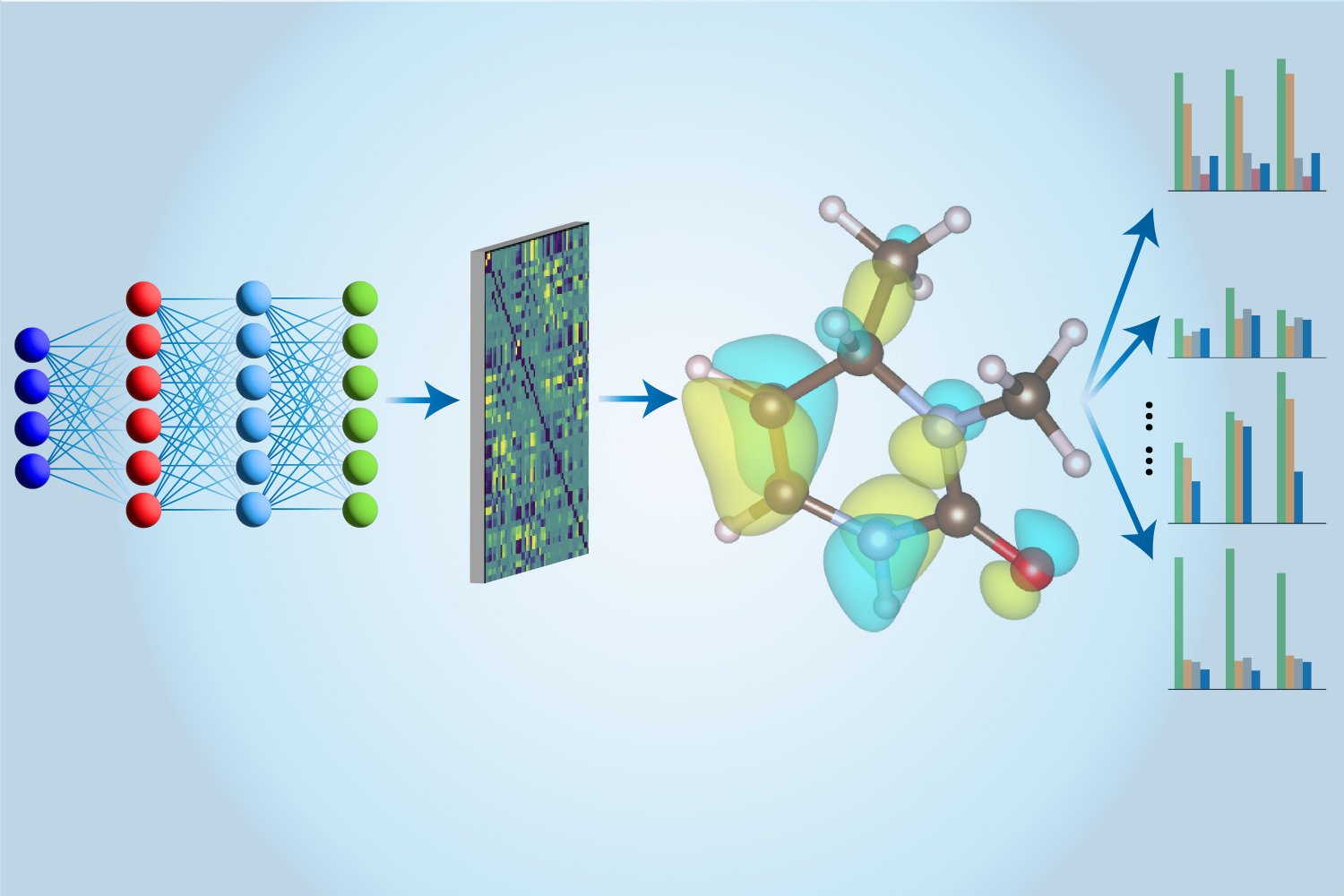
研究人员利用机器学习和贝叶斯优化框架,成功实现了对含镓材料的反向设计,生成了可调带隙(0.5–3.5 eV)的新型半导体。这一方法加速了材料发现,具有重要的光电和能量转换应用潜力,且生成材料具有100%的新颖性,为未来材料设计提供了新的思路和工具。
本研究提出了一种新框架,结合MatSciBERT的实体提取模型与指针网络和注意力机制,从科学文献中提取多元组信息。实验结果表明,该模型在多个数据集上表现优异,有效支持材料设计和数据驱动创新。
本研究提出了一种结合大型语言模型与动态图表示的自主图扩展框架,克服了传统知识图谱的局限性。该系统通过迭代生成新概念与关系,构建可持续的知识网络,展示了在材料设计等领域的应用潜力。
DARWIN 1.5模型通过自然语言处理和多任务学习,显著提高了材料属性预测的准确性,超越了传统方法。该模型整合了大量科学问答数据,优化了材料设计,展现出在材料科学领域的广泛应用潜力。
MatterGen是微软研究院开发的生成式AI材料设计工具,能够高效生成新材料,克服传统材料发现的局限。通过定制化的扩散模型和适配器模块,MatterGen生成的无机材料在稳定性和多样性上显著优于现有方法,实验验证了其实际应用潜力。
研究人员结合量子化学的耦合簇理论与机器学习,开发了新模型MEHnet,能够更快速、准确地预测分子特性。这一方法有望在材料设计中实现高通量筛选,促进新材料的发现与应用。
麻省理工学院4-061实验室研究固态聚合物电解质锂电池,利用人工智能和机器学习推动创新。与丰田研究所合作,研究团队通过生成式AI技术设计新型聚合物,提升电池材料性能,展示了AI在材料设计中的潜力。
量子计算机有望革新药物发现和材料设计,但其可靠性仍需提升。AlphaQubit是一种基于AI的解码器,能够准确识别量子计算错误,结合了Google DeepMind和Google Quantum AI的技术。通过冗余和一致性检查,AlphaQubit在解码准确性上超越了现有方法,为量子计算的实用化奠定基础。
研究团队提出了统一模型UniIF,成功解决了分子逆折叠中的三大挑战,适用于蛋白质、RNA和材料设计。实验结果显示,UniIF在各项任务中表现优异,超越现有方法,具有广泛的应用潜力。
本文探讨了利用晶体图神经网络(CGNN)和机器学习技术预测高熵合金的材料属性。研究提出了数值推理方法和图神经网络模型,以解决材料设计中的假设局限性,推动人工智能在材料发现中的应用。通过构建知识图谱和多智能体系统,提升了材料设计的准确性和效率。
本文探讨了深度强化学习在分子设计中的应用,提出了基于球谐级数的三维分子设计、分层代理方法和生成模型等新策略,显著提高了分子生成的效率和多样性。这些方法在药物发现和材料设计中展现出优越性,推动了计算化学的发展。
本文介绍了一种基于扩散模型和机器学习的新型晶体结构生成方法,能够生成稳定的无机材料。该方法通过学习材料数据分布,优化特定性质,显著提高了晶体结构预测的准确性和效率,推动了材料设计的进展。
本研究提出了超图神经网络(HGNN)和分子超图神经网络(MHNN),结合高通量计算和机器学习,旨在高效处理复杂数据和预测材料性质。实验结果表明,这些方法在纳米颗粒分割和材料属性预测方面表现优异,推动了自动化材料设计的发展。
本文探讨了在深空环境中设计具有灵活物理化学性质的材料的重要性,提出了多种基于机器学习的方法,如神经介质网络、深度神经网络和随机森林,以优化材料设计,尤其是在数据稀缺的情况下。这些方法能够快速生成具有特定性能的变形材料,推动超材料的研究与应用。
斯坦福大学和美国能源部 SLAC 国家加速器实验室的科学家们开发了一种 AI 方法,能够更有效地收集数据,以应对复杂的材料设计挑战。研究人员提出了一个框架,通过用户定义的过滤算法来捕获实验目标,并自动转换为智能的数据采集策略。该方法在实验中证明了高效性,可快速发现新材料,对气候变化、量子计算和药物设计等领域有潜在应用。
该文章介绍了将深度学习与密度泛函理论相结合的新材料设计方法,通过神经网络预测材料的电子结构和性质。研究人员构建了一个包含104种固体材料的大型数据库,并成功开发了一个通用材料模型。该模型通过训练和微调能够准确预测材料的能带结构和其他性质。这种深度学习方法为创新材料发现提供了新机遇,但仍面临一些挑战。
本文探讨了机器学习与过程挖掘结合的关键问题,提出了整体优化方法以促进材料设计与工艺设计的结合。研究展示了强化学习和多任务学习在金属成型过程中的应用,强调了新数字框架在化学行业中的潜力,以及无监督故障检测方法的有效性。
本文介绍了一种基于主动学习算法的全自动方法,用于生成分子能量数据集,以支持深度学习算法在有机分子中的能量和力预测。研究表明,主动学习显著节约标注和计算成本,并在材料设计和药物发现中提供高效筛选方法。文章还探讨了机器学习在量子模拟和科学研究中的应用潜力。
该研究提出了一种结合势能和机器学习的模型,能够准确计算小分子与生物分子间的非共价相互作用。通过机器学习内原子势函数(MLIP)框架,研究探讨了经典与机器学习势能的边界,强调了全面训练数据在自由能预测中的重要性,并展示了机器学习势能在材料设计中的应用潜力。
本文探讨了机器学习在化学和聚烯烃制造中的应用,重点在材料设计与工艺优化。研究提出结合强化学习和多任务学习的整体优化方法,解决材料和工艺设计问题,并介绍了基于模式的无监督纺织品异常检测方法,展示了机器学习在提高生产效率和降低成本方面的潜力。
完成下面两步后,将自动完成登录并继续当前操作。