
新的机器学习应用程序帮助研究人员预测化学属性
内容提要
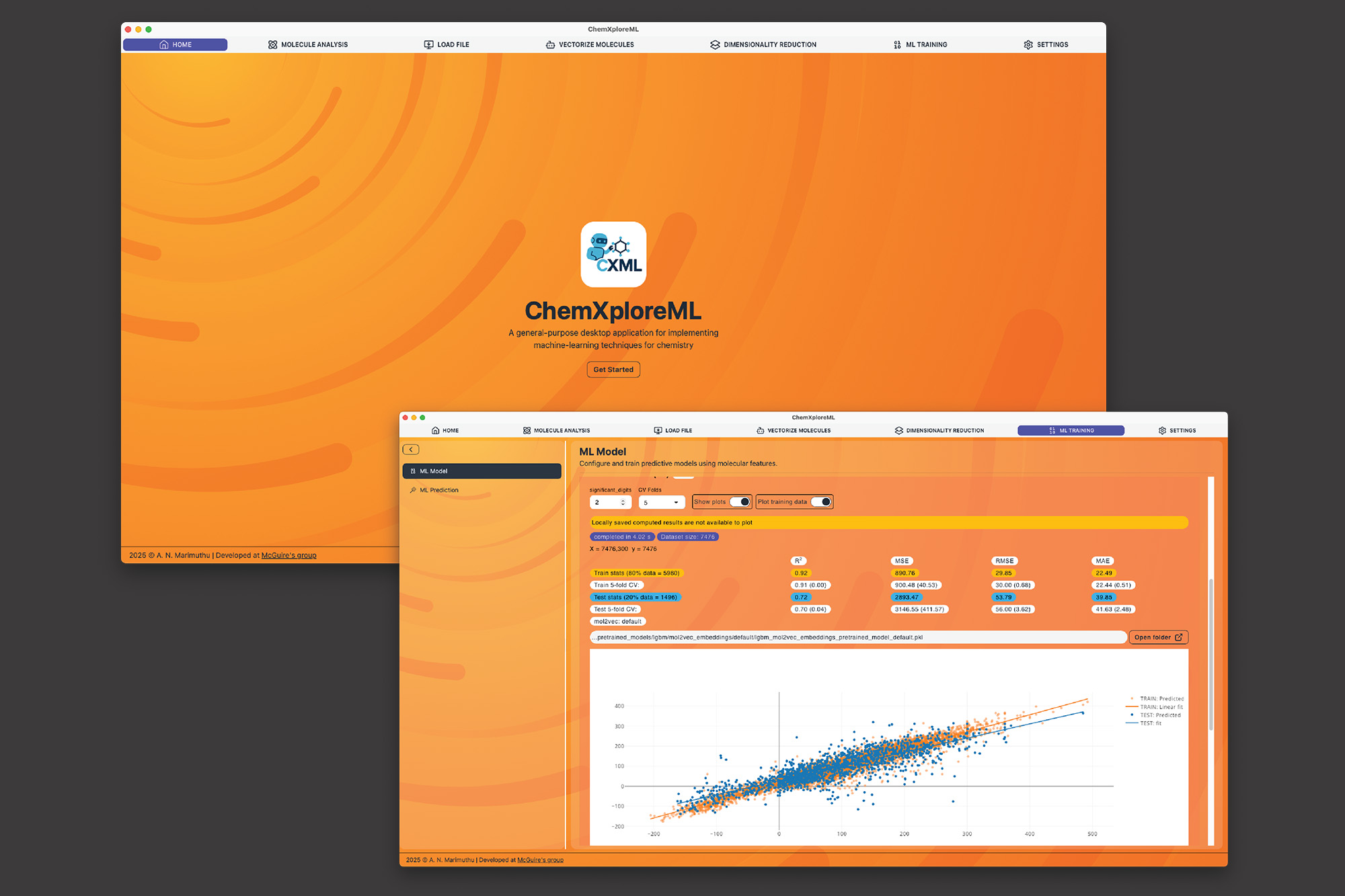
麻省理工学院的研究人员开发了ChemXploreML,这是一款用户友好的桌面应用程序,利用机器学习预测分子属性,无需深厚的编程技能。该应用程序能够自动将分子结构转化为数值语言,并通过直观界面快速预测熔点、沸点等属性,旨在加速新药和材料的研发。ChemXploreML可离线使用,确保研究数据的保密性,并具备未来技术的整合能力。
关键要点
-
麻省理工学院的研究人员开发了ChemXploreML,这是一款用户友好的桌面应用程序,利用机器学习预测分子属性。
-
ChemXploreML能够自动将分子结构转化为数值语言,并通过直观界面快速预测熔点、沸点等属性。
-
该应用程序旨在加速新药和材料的研发,降低研究成本,提高预测效率。
-
ChemXploreML可离线使用,确保研究数据的保密性,并具备未来技术的整合能力。
-
该应用程序在有机化合物的五个关键分子属性上进行了测试,准确率高达93%。
延伸解读
机器学习在化学研究中的应用前景
ChemXploreML的推出标志着机器学习在化学领域的应用进入了一个新阶段。通过简化分子属性预测的过程,研究人员能够更快地进行药物和材料的开发。这种技术的普及将使更多化学家能够利用先进的预测模型,推动科学研究的进展。
数据保密性的重要性
ChemXploreML的离线功能确保了研究数据的保密性,这在化学研究中尤为重要。许多研究涉及敏感数据,离线操作可以有效防止数据泄露,保护知识产权。这一设计考虑了研究人员的实际需求,增强了应用的实用性。
未来技术的整合能力
ChemXploreML的设计允许未来技术和算法的无缝整合,这为研究人员提供了持续更新的可能性。随着机器学习技术的不断进步,ChemXploreML将能够适应新的研究需求,保持其在化学预测领域的竞争力。
延伸问答
ChemXploreML是什么?
ChemXploreML是一款由麻省理工学院研究人员开发的桌面应用程序,利用机器学习预测分子属性,用户无需深厚的编程技能。
ChemXploreML如何帮助化学研究?
ChemXploreML通过自动将分子结构转化为数值语言,快速预测熔点、沸点等属性,从而加速新药和材料的研发。
ChemXploreML的准确性如何?
该应用程序在有机化合物的五个关键分子属性上进行了测试,准确率高达93%。
ChemXploreML的离线功能有什么优势?
ChemXploreML可离线使用,确保研究数据的保密性,避免数据泄露。
ChemXploreML如何降低研究成本?
通过提供快速和高效的分子属性预测,ChemXploreML降低了传统方法所需的时间和设备成本。
ChemXploreML的未来发展方向是什么?
ChemXploreML设计为可随时间演变,未来技术和算法可以无缝整合,确保研究人员使用最新的方法。
