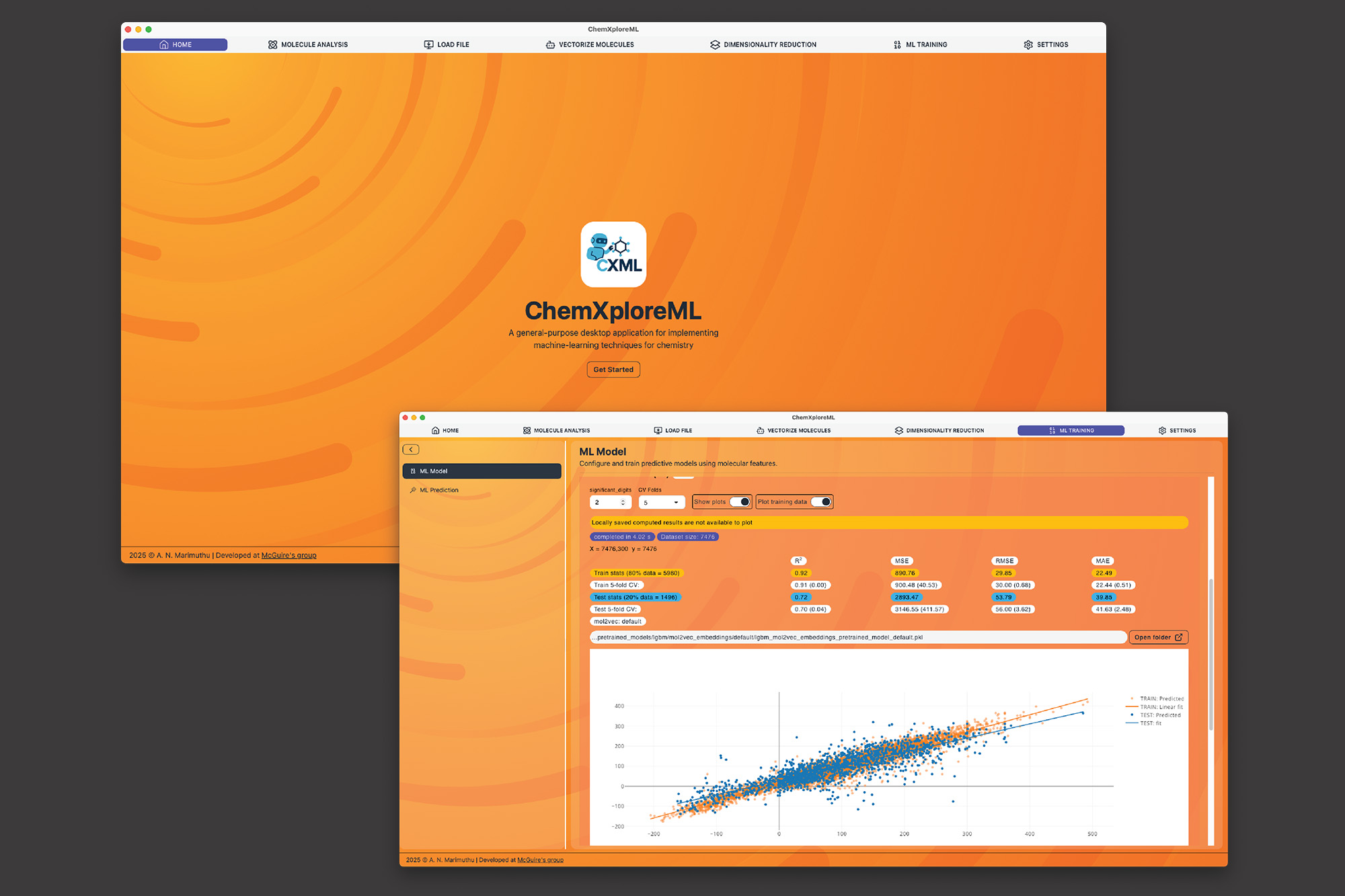
麻省理工学院的研究人员开发了ChemXploreML,这是一款用户友好的桌面应用程序,利用机器学习预测分子属性,无需深厚的编程技能。该应用程序能够自动将分子结构转化为数值语言,并通过直观界面快速预测熔点、沸点等属性,旨在加速新药和材料的研发。ChemXploreML可离线使用,确保研究数据的保密性,并具备未来技术的整合能力。
本研究探讨了图学习在药物设计和分子属性预测中的基准挑战,指出现有基准缺乏实际应用的关注,建议采用更具意义的基准和评估协议,以促进研究进展和领域合作。
完成下面两步后,将自动完成登录并继续当前操作。